 订阅
订阅Human genetic diversity: Lewontin’s fallacy
人类遗传多样性之列万廷的谬误
作者:A.W.F. Edwards
翻译:小聂(@PuppetMaster)
校对:辉格(@whigzhou)
来源:Edwards, A. W. F. (2003). “Human genetic diversity: Lewontin’s fallacy”. BioEssays 25 (8): 798–801.
Summary
In popular articles that play down the genetical differences among human populations, it is often stated that about 85% of the total genetical variation is due to individual differences within populations and only 15% to differences between populations or ethnic groups. It has therefore been proposed that the division of Homo sapiens into these groups is not justified by the genetic data. This conclusion, due to R.C. Lewontin in 1972, is unwarranted because the argument ignores the fact that most of the information that distinguishes populations is hidden in the correlation structure of the data and not simply in the variation of the individual factors. The underlying logic, which was discussed in the early years of the last century, is here discussed using a simple genetical example.
概要
在那些淡化人类种群遗传差异的流行文章里面,一个常见的说法是:85%的遗传差异来自于种群内的个体间差异,而只有15%是来自于种群或种族间差异。因此,有人认为依靠遗传数据而将智人划分为不同群体,是不合理的。R.C.列万廷在1972年作出的该论断是缺乏根据的,原因在于:用于区分种群的大部分信息隐藏在遗传数据的相关性结构里,而不简简单单体现在单个因子的差异上。这背后的逻辑,在上世纪初就已被讨论,在这里用一个简单的遗传学范例来加以说明。
“When a large number of individuals [of any kind of organism] are measured in respect of physical dimensions, weight, colour, density, etc., it is possible to describe with some accuracy the population of which our experience may be regarded as a sample. By this means it may be possible to distinguish it from other populations differing in their genetic origin, or in environmental circumstances. Thus local races may be very different as populations, although individuals may overlap in all characters; . . .” R.A. Fisher (1925).
“在测量「任何一种有机体的」大量个体的物理属性——重量,颜色,密度等——的时候,我们可以以特定的精确度来对该有机体的种群加以描述,虽然在经验上,他们可能只是样本而已。用这种方式,我们有可能把他们和其他种群在遗传起源上,甚至是在环境条件上,加以区分。这样一来,地区亚种之间在种群层面上可以有巨大的差别,尽管个体之间有可能高度重合……”R.A.菲舍尔(1925)。
“It is clear that our perception of relatively large differences between human races and subgroups, as compared to the variation within these groups, is indeed a biased perception and that, based on randomly chosen genetic differences, human races and populations are remarkably similar to each other, with the largest part by far of human variation being accounted for by the differences between individuals. Human racial classification is of no social value and is positively destructive of social and human relations. Since such racial classification is now seen to be of virtually no genetic or taxonomic significance either, no justification can be offered for its continuance”. R.C. Lewontin (1972).
“很明显,我们对于人类种群或是亚种之间差异大于群内个体差异的理解,是一种偏见。并且,基于随机选定的遗传差异来看,人类种族和种群之间具有显著的相似性,迄今为止人类间差异的最大部分都源于个体差异。种族分类不仅没有社会价值,而且对人和社会的关系有着强烈的破坏性。既然现在看来这样的种族分类毫无遗传学和分类学根据,将其持续下去也是毫无必要的了。”R.C.列万廷(1972)。
“The study of genetic variations in Homo sapiens shows that there is more genetic variation within populations than between populations. This means that two random individuals from any one group are almost as different as any two random individuals from the entire world. Although it may be easy to observe distinct external differences between groups of people, it is more difficult to distinguish such groups genetically, since most genetic variation is found within all groups.” Nature (2001).
“对于智人遗传差异的研究表明种群内差异大于种群间差异。这意味着从同一族群中随机挑选的两个个体之间的差异几乎等同于世界上任何两个随机个体间的差异。尽管我们可以观测到族群间分明的外部特征区别,对他们在遗传上加以区分却困难得多,因为大部分遗传差异存在于所有的族群之内。”《自然》(2001)。
Introduction
导言
In popular articles that play down the genetical differences among human populations it is often stated, usually without any reference, that about 85% of the total genetical variation is due to individual differences within populations and only 15% to differences between populations or ethnic groups. It has therefore been suggested that the division of Homo sapiens into these groups is not justified by the genetic data. People the world over are much more similar genetically than appearances might suggest.
淡化人类种群间遗传差异的流行文章里,一个常见的未加引用的说法是:85%的遗传差异源于种群内的个体间差异,而只有15%的差异来自于种群或是种族。因此以这些种群为智人分类是不被遗传数据支持的。世界各地的人们在遗传上的相似性远大于外表所显示出的那样。
Thus an article in New Scientist reported that in 1972 Richard Lewontin of Harvard University “found that nearly 85 per cent of humanity’s genetic diversity occurs among individuals within a single population.”“In other words, two individuals are different because they are individuals, not because they belong to different races.” In 2001, the Human Genome edition of Nature came with a compact disc containing a similar statement, quoted above.
正如《新科学家》的一篇文章所报道的,哈佛大学的理查德•列万廷在1972年“发现近85%的人类遗传差异产生于种群内的个体之间”、“换句话说,个体之所以不同是因为他们是不同的个体,而不是因为他们属于不同的种族。”2001年,《自然:人类基因组特刊》附带的压缩光盘内也包含类似的引述。
Such statements seem all to trace back to a 1972 paper by Lewontin in the annual review Evolutionary Biology. Lewontin analysed data from 17 polymorphic loci, including the major blood-groups, and 7 ‘races’ (Caucasian, African, Mongoloid, S. Asian Aborigines, Amerinds, Oceanians, Australian Aborigines). The gene frequencies were given for the 7 races but not for the individual populations comprising them, although the final analysis did quote the within-population variability.
类似的陈述貌似都出自列万廷在1972年《进化生物学》年度综述中发表的一篇文章。列万廷分析了出自17个多态基因位点(包括主要的血型)和7个“种族”(高加索人,非洲人,蒙古人,南亚原住民,美洲印第安人,大洋洲人,以及澳洲原住民)的数据。尽管最终的分析引述了种群内多样性,对于基因频率,文章只给出了7个种族的数据,而没有给出组成这些种族的种群数据。
“The results are quite remarkable. The mean proportion of the total species diversity that is contained within populations is 85.4%…. Less than 15% of all human genetic diversity is accounted for by differences between human groups! Moreover, the difference between populations within a race accounts for an additional 8.3%, so that only 6.3% is accounted for by racial classification.”
“结果很显著。在种群内包含的总物种多样性的比例平均可以达到85.4%……而只有不到15%的人类遗传多样性能被种群差异所解释!不仅如此,同一种族内部种群的差异在这里占8.3%,所以只有6.3%的差异能归结于种族划分。”
Lewontin concluded “Since . . . racial classification is now seen to be of virtually no genetic or taxonomic significance . . ., no justification can be offered for its continuance” (full quotation given above).
列万廷结论道:“既然如此……种族划分现在看来毫无遗传学或是分类学依据……延续它看来是毫无必要的”(上文已有完整引用)。
Lewontin included similar remarks in his 1974 book The Genetic Basis of Evolutionary Change “The taxonomic division of the human species into races places a completely disproportionate emphasis on a very small fraction of the total of human diversity. That scientists as well as nonscientists nevertheless continue to emphasize these genetically minor differences and find new ‘scientific’ justifications for doing so is an indication of the power of socioeconomically based ideology over the supposed objectivity of knowledge.”
在他1974年的书《进化改进的遗传学基础》种,列万廷加入了类似的评论。“将人类划分为种族的分类学方法将不成比例的注意力集中在人类总体多样性的零头上。科学家和大众对这种零星遗传差异的重视和坚持,甚至发现新的‘科学’证据来支持这种做法,显示出社会经济学意识形态的力量超越了本该是客观的知识。”
The fallacy
谬误
These conclusions are based on the old statistical fallacy of analysing data on the assumption that it contains no information beyond that revealed on a locus-by-locus analysis, and then drawing conclusions solely on the results of such an analysis. The ‘taxonomic significance’ of genetic data in fact often arises from correlations amongst the different loci, for it is these that may contain the information which enables a stable classification to be uncovered.
之所以有以上定论,主要是出于一个陈旧的统计谬误,即认为除了基于单个位点的分析结论之外,数据不包含任何其它信息,并且只考虑基于这个假设的分析和它衍生出的结论。而所谓的“有分类学意义”的遗传数据实际上源自不同位点之间的相关关系,正是这些相关关系中可能包含的信息驱动了对于分类的发掘。
Cavalli-Sforza and Piazza coined the word ‘treeness’ to describe the extent to which a tree-like structure was hidden amongst the correlations in gene-frequency data. Lewontin’s superficial analysis ignores this aspect of the structure of the data and leads inevitably to the conclusion that the data do not possess such structure. The argument is circular. A contrasting analysis to Lewontin’s, using very similar data, was presented by Cavalli-Sforza and Edwards at the 1963 International Congress of Genetics. Making no prior assumptions about the form of the tree, they derived a convincing evolutionary tree for the 15 populations that they studied. Lewontin, though he participated in the Congress, did not refer to this analysis.
Cavalli-Sforza和 Piazza创造了“树性”这一词汇,用于描述一个树形结构在基因频率数据的相关关系中的隐匿程度。列万廷的肤浅分析无视了数据在这方面的特性,于是不可避免的得出结论认为该树形结构不存在。这是个循环论证。Cavalli-Sforza和Edwards于1963年的世界遗传大会发表了与之对应的对比分析,并使用了类似的数据。在不对树形做任何先验假设的情况下,他们在研究的15个种群中得出了一个令人信服的进化树结构。列万廷虽然参加了此次会议,但却没有提到这个分析。
The statistical problem has been understood at least since the discussions surrounding Pearson’s ‘coefficient of racial likeness’ in the 1920s. It is mentioned in all editions of Fisher’s Statistical Methods for Research Workers from 1925 (quoted above). A useful review is that by Gower in a 1972 conference volume The Assessment of Population Affinities in Man. As he pointed out, “…the human mind distinguishes between different groups because there are correlated characters within the postulated groups.”
早在围绕皮尔森在1920年代提出的“种族相似性的协同因素”的讨论中,人们就已经理解了相关的统计原理了。在菲舍尔所作的《给研究员的统计方法》的所有版本中,该原理都有被提及(本文开头亦有引用)。高尔于1972年在“人类种群亲缘关系评估”的会议出版物中提出了一个有用的评论,他指出:“……人类心智将人划分为不同的组别,原因在于在这些组别内存在具有相关性的特性。”
The original discussions involved anthropometric data, but the fallacy may equally be exposed using modern genetic terminology. Consider two haploid populations each of size n. In population 1 the frequency of a gene, say ‘+’ as opposed to ‘-’, at a single diallelic locus is p and in population 2 it is q, where p + q = 1. (The symmetry is deliberate.) Each population manifests simple binomial variability, and the overall variability is augmented by the difference in the means.
原初的讨论涉及一些人体测量学数据,但是我们用现代遗传学术语也同样可以揭示这个谬误。考虑两个个体数量各为n的单倍体种群。在种群1中某基因在一个单独位点为“+”而不是“-”的频率为p,在种群2中该频率为q,且p + q = 1。(这种对称性是有意设定的。)各种群的多样性为简单二项式分布,且总体多样性由于两个种群间平均值的差异而得到加强。
The natural way to analyse this variability is the analysis of variance, from which it will be found that the ratio of the within-population sum of squares to the total sum of squares is simply 4pq. Taking p = 0.3 and q = 0.7, this ratio is 0.84; 84% of the variability is within-population, corresponding closely to Lewontin’s figure. The probability of misclassifying an individual based on his gene is p, in this case 0.3. The genes at a single locus are hardly informative about the population to which their bearer belongs.
很自然的,我们用方差分析来评估多样性,从中可以得出种群内平方和与总体平方和之比为4pq【译注:对于任一种群,种群方差为npq,种群平方和为n2pq;总和平方和为1/4•n2(p+q)2 = 1/4•n2;(n2pq)/( 1/4•n2)=4pq】。如 p = 0.3 而 q = 0.7,该比率为0.84,即84%的多样性来自于种群内,正好对应列万廷的结果。基于该基因对个体的分类误差率为p,即0.3。单个位点的基因几乎不包含关于该基因携带者属于哪个种群的任何信息。
Now suppose there are k similar loci, all with gene frequency p in population 1 and q in population 2. The ratio of the within-to-total variability is still 84% at each locus. The total number of ‘+’ genes in an individual will be binomial with mean kp in population 1 and kq in population 2, with variance kpq in both cases. Continuing with the former gene frequencies and taking k = 100 loci (say), the mean numbers are 30 and 70 respectively, with variances 21 and thus standard deviations of 4.58. With a difference between the means of 40 and a common standard deviation of less than 4.6, there is virtually no overlap between the distributions, and the probability of misclassification is infinitesimal, simply on the basis of counting the number of ‘+’ genes. Fig. 1 shows how the probability falls off for up to 20 loci.
现在假设共有k个相似位点,都在种群1中和种群2中分别具有p和q的基因频率。在每个单个位点上,种群内多样性与总体多样性之比仍是84%。在每个个体上为“+”的基因数将呈二项式分布,其均值在种群1中为kp,在种群2中为kq,方差在两个种群中同为kpq。继续之前关于基因频率的假设【译注:即p = 0.3,q = 0.7】,设k = 100 个位点,则在两个种群中均值各为30和70,方差为21,因此标准差为4.58。在均值相差40的情况下,共同的标准差还不到4.6,因此这两个分布几乎没有任何重叠部分,所以基于“+”基因出现个数所作分类的误差可能性是无限小。图1显示了该分类误差率随位点数增加而下降的曲线,至20个位点。
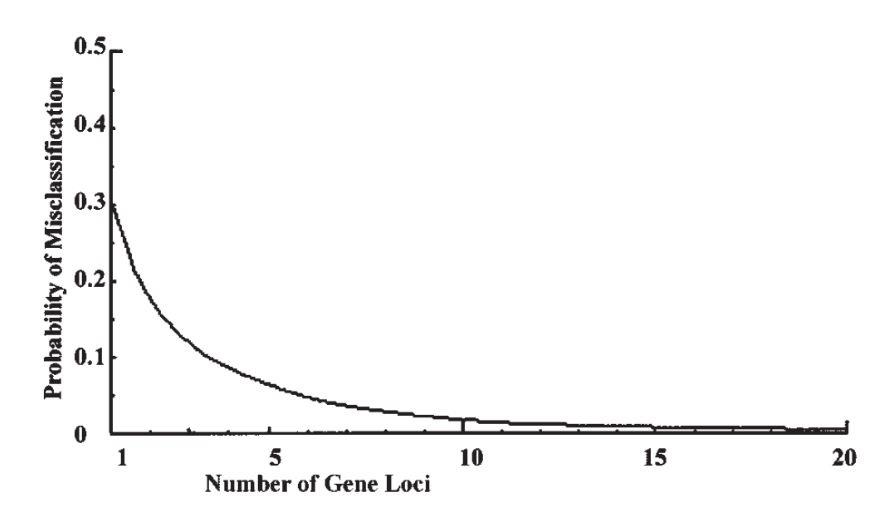
Figure 1. Graph showing how the probability of misclassification falls off as the number of gene loci increases, for the first example given in the text. The proportion of the variability within groups remains at 84% as in Lewontin’s data, but the probability of misclassification rapidly becomes negligible.
图1. 该图显示分类误差率随基因位点数增加而下降的曲线,用于文本中第一个例证。组内多样性占比依旧为列万廷数据揭示的84%,但分类误差率迅速下降至可以忽略的程度。【图表横轴:基因位点数;图表纵轴:分类误差率】
One way of looking at this result is to appreciate that the total number of ‘+’ genes is like the first principal component in a principal component analysis (Box 1). For this component the between-population sum of squares is very much greater than the within-population sum of squares. For the other components the reverse will hold, so that overall the between-population sum of squares is only a small proportion (in this example 16%) of the total. But this must not beguile one into thinking that the two populations are not separable, which they clearly are.
一种领会该结果的方式是将“+”基因的总数看成主成分分析法中的第一主成分(见框文1【编注:是对主成分分析(Principal components analysis,PCA)方法的介绍,译略,有兴趣可查看原文,或参见维基词条“主成分分析”】)。对于该成分,种群间平方和远大于种群内平方和。对于其他成分则反之,以至于对所有成分来说种群间平方和仅占总体平方和的一小部分(在这个例子里面为16%)。但这个结果不能诱使我们认为两个种群是不可分的,而实际上他们是清晰可分的。
Each additional locus contributes equally to the within-population and between-population sums of squares, whose proportions therefore remain unchanged but, at the same time, it contributes information about classification which is cumulative over loci because their gene frequencies are correlated.
每一个增加的位点都同样的增加种群内和种群间的平方和,导致它们之间的比率不变。但同时,关于分类的信息也增加了,而且这种增加在位点数量上是具有累加性的,因为位点之间的基因频率是相关的。
Classification
分类
It might be supposed, though it would be wrong, that this example is prejudiced by the assumptions that membership of the two populations is known in advance and that, at each locus, it is the same population that has the higher frequency of the ‘+’ gene. In fact the only advantage of the latter simplifying assumption was that it made it obvious that the total number of ‘+’ genes is the best discriminant between the two populations.
人们或可认为——虽然这么想是错的——这是个不太好的例子,因为假设了个体在两个种群的归属事先已知,并且在每个位点都是同一种群拥有较高的获得“+”的基因频率。实际上,后一个简化假设的唯一优势在于使得“+”基因的总数成为种群的明显最优判准。
To dispel these concerns, consider the same example but with ‘+’ and ‘-’ interchanged at each locus with probability 1⁄2, and suppose that there is no prior information as to which population each individual belongs. Clearly, the total number of ‘+’ genes an individual contains is no longer a discriminant, for the expected number is now the same in each group. A cluster analysis will be necessary in order to uncover the groups, and a convenient criterion is again based on the analysis of variance as in the method introduced by Edwards and Cavalli-Sforza. Here the preferred division into two clusters maximises the between-clusters sum of squares or, what is the same thing, minimises the sum of the within-clusters sums of squares.
为解除这些疑虑,设想同样的例子,但“+”和“-”在各个位点以1/2的概率互换,且没有关于个体归属的任何先验信息。显然,个体所拥有的“+”基因总数不能再作为判准,因为该数目的期望值在两组里面是一样的。在这种情况下需要用聚类分析来处理分组,且一个便利的分组条件仍然是基于方差分析的,其方法由Edwards和Cavalli-Sforza提供。这里对于聚类的优先分割会最大化聚类间平方和,或者说是最小化聚类内平方和,在这里是一样的意思。
As pointed out by these authors, it is extremely easy to compute these sums for binary data, for all the information is contained in the half-matrix of pairwise distances between the individuals, and at each locus this distance is simply 0 for a match and 1 for a mismatch of the genes. Since interchanging ‘+’ and ‘-’ makes no difference to the numbers of matches and mismatches, it is clear that the random changes introduced above are irrelevant.
正如这两位作者指出的那样,在二值数据里是很容易计算出这些平方和的,因为所有信息都可以体现为一个成对个体间的距离半矩阵。在每个位点上,配对时距离为0,不配对为1。既然互换“+”和“-”对于配对关系没有影响,那么显然以上引入的随机变化是无关的。
Continuing the symmetrical example, the probability of a match is p2 + q2 if the two individuals are from the same population and 2pq if they are from different populations. With k loci, therefore, the distance between two individuals from the same population will be binomial with mean k(p2 + q2) and variance k(p2 + q2)(1 – p2 – q2) and if from different populations binomial with mean 2kpq and variance 2kpq(1 – 2pq). These variances are, of course, the same.
继续这个对称性例子,对于来自同一种群的两个个体来说,单一位点配对的机率为p2 + q2;若来自不同种群,则为2pq。因此,对于k个位点,同一种群两个个体间距离呈二项式分布,均值为k(p2 + q2),方差为k(p2 + q2)(1 – p2 – q2);若来自不同种群,则均值为2kpq,方差为2kpq(1 – 2pq)。这两个方差显然是一样的【译注:p + q = 1 à p2 + q2 = 1 – 2pq】。
Taking p = 0.3, q = 0.7 and k = 100 as before, the means are 58 and 42 respectively, a difference of 16, the variances are 24.36 and the standard deviations both 4.936. The means are thus more than 3 standard deviations apart (3.2415). The entries of the half-matrix of pairwise distances will therefore divide into two groups with very little overlap, and it will be possible to identify the two clusters with a risk of misclassification which tends to zero as the number of loci increases.
像之前一样,取p = 0.3, q = 0.7 和 k = 100,则均值分别为58和42,相差16。方差为24.36,即两组的标准差都为4.936。这样一来两组均值之间则有超出3个标准差的距离。因此,这个成对个体距离半矩阵中的数值就可以被分成几乎没有重叠的两组,这样就有可能以较小的分类误差来识别两个聚类,且该分类误差率随位点数目增加逐渐趋向于0。
By analogy with the above example, it is likely that a count of the four DNA base frequencies in homologous tracts of a genome would prove quite a powerful statistical discriminant for classifying people into population groups.
同理可知,对于基因组同源区域的四个DNA碱基频率进行计数,这种方法很可能被证明是一个十分有效的进行种群分类的统计判准。
Conclusion
结论
There is nothing wrong with Lewontin’s statistical analysis of variation, only with the belief that it is relevant to classification. It is not true that “racial classification is . . . of virtually no genetic or taxonomic significance”. It is not true, as Nature claimed, that “two random individuals from any one group are almost as different as any two random individuals from the entire world”, and it is not true, as the New Scientist claimed, that “two individuals are different because they are individuals, not because they belong to different races” and that “you can’t predict someone’s race by their genes”. Such statements might only be true if all the characters studied were independent, which they are not.
列万廷对于多样性的统计分析本身没错,错的是认为该分析与分类有关。那种认为“人种分类……毫无遗传学和分类学依据”的想法是错误的。类似《自然》杂志所声明的“两个来源于任何组别的个体之间的差异和两个来源于世界上任何地方的个体之间的差异几乎一样大”的想法是错误的。类似《新科学家》声明的“两个个体有区别是因为他们是两个个体,而不是因为他们从属于不同的种族”和“你无法通过基因来判断某人的种族”的想法也是错误的。这类声明唯有在所有被研究的特性都是独立分布的时候才成立,可它们并不是独立分布的。
Lewontin used his analysis of variation to mount an unjustified assault on classification, which he deplored for social reasons. It was he who wrote “Indeed the whole history of the problem of genetic variation is a vivid illustration of the role that deeply embedded ideological assumptions play in determining scientific ‘truth’ and the direction of scientific inquiry”.
列万廷利用他对于多样性的分析对人种分类发起了一场毫无根据的攻击,认为人种分类是社会因素造成的悲剧。正是他写道:“的确,遗传多样性问题的整个研究历史生动的向我们展示了深埋的意识形态假设是如何决定科学的‘真相’和科学探索的方向。”
In a 1970 article Race and intelligence he had earlier written “I shall try, in this article, to display Professor Jensen’s argument, to show how the structure of his argument is designed to make his point and to reveal what appear to be deeply embedded assumptions derived from a particular world view, leading him to erroneous conclusions.”
更早之前在一篇发表于1972年的题为“种族与智能”的文章里他写道:“我会努力在本文向你展示詹森教授的论据,以及他如何构筑这些论据以说明他的论点的,并向你揭示那些深埋的假设是如何来自于一种特别的世界观,最终导致他得出一个错误的结论。”
A proper analysis of human data reveals a substantial amount of information about genetic differences. What use, if any, one makes of it is quite another matter. But it is a dangerous mistake to premise the moral equality of human beings on biological similarity because dissimilarity, once revealed, then becomes an argument for moral inequality. One is reminded of Fisher’s remark in Statistical Methods and Scientific Inference “that the best causes tend to attract to their support the worst arguments, which seems to be equally true in the intellectual and in the moral sense.”
对于人类数据的恰当分析揭示了有关遗传差异的大量信息。如何利用这些信息则是另外一回事。但将生物学上的相似性当作人类在道德上平等的前提是一个危险的错误,因为差异一旦被发现,就会被视作道德不平等的论据。我们应当铭记菲舍尔在《统计方法与科学推断》中的话:“最好的主张常常会吸引最差的理由,而这理由在智力上和道德上都一样不靠谱。”
Epilogue
后记
This article could, and perhaps should, have been written soon after 1974. Since then many advances have been made in both gene technology and statistical computing that have facilitated the study of population differences from genetic data. The magisterial book of Cavalli-Sforza, Menozzi and Piazza took the human story up to 1994, and since then many studies have amply confirmed the validity of the approach.
这篇文章本可以——且应该——在1974年之后不久就写完。自那时起,基因技术和统计计算方面取得了诸多进展,为研究遗传数据中的种群差异提供了很大帮助。Cavalli-Sforza, Menozzi和Piazza的权威著作将人类的故事带到了1994年,从那时起,众多研究广泛的证实了他们的方法。
Very recent studies have treated individuals in the same way that Cavalli-Sforza and Edwards treated populations in 1963, namely by subjecting their genetic information to a cluster analysis thus revealing genetic affinities that have unsurprising geographic, linguistic and cultural parallels. As the authors of the most extensive of these comment, “it was only in the accumulation of small allele-frequency differences across many loci that population structure was identified.”
一些晚近的研究用Cavalli-Sforza和Edwards处理种群的方法对待个体,即,将聚类分析运用到个体遗传信息的分析上,由此所揭示出的地理、语言、以及文化上的遗传亲缘性,并不出乎意料。正如在这些课题上之中涉猎最为广泛的作者所言:“只有当众多微小位点上等位基因的差异逐渐累积,种群结构才得以显现。”
(编辑:辉格@whigzhou)
*注:本译文未经原作者授权,本站对原文不持有也不主张任何权利,如果你恰好对原文拥有权益并希望我们移除相关内容,请私信联系,我们会立即作出响应。
——海德沙龙·翻译组,致力于将英文世界的好文章搬进中文世界——
本文不开放评论